Users of the standard IEC 60601-2-24:2012 (infusion pumps and controllers) might scratch their heads over some of the requirements and details associated with performance tests. To put it bluntly, many things just don’t make sense.
A recent project comparing the first and second edition along with the US version AAMI ID26 offers some possible clues as to what happened.
The AAMI standard, formally ANSI/AAMI ID26 is a modified version of IEC 60601-2-24:1998. It includes some dramatic deviations and arguably makes performance testing significantly more complex. This AAMI standard has since been withdrawn and is no longer available for sale, and is not used by the FDA as a recognized standard. But the influence on the 2012 edition of IEC 60601-2-24 is such that it is worth getting a copy of this standard if you can, just to understand what is going on.
It seems that the committee working on the new edition of IEC 60601-2-24 were seriously looking at incorporating some of the requirements from the AAMI standard, and got as far as deleting some parts, adding some definitions and then … forgot about it and published the standard anyway. The result is a standard that does not make sense in several areas.
For example, in IEC 60601-2-24:1998 the sample interval for general performance tests is fixed at 0.5 min. In the AAMI edition, this is changed to 1/6min (10 sec). In IEC 60601-2-24:2012, the sample interval is ... blank. It’s as if the authors deleted the text “0.5 min”, were about to type in “1/6 min”, paused to think about it, and then just forgot. Maybe there was a lot of debate about the AAMI methods, which are indeed problematic, followed by huge pressure to get the standard published so that it could be used with the 3rd edition of IEC 60601-1, so they just hit the print button without realising some of the edits were only half baked. Which we all do, but … gee … this is an international standard used in a regulatory context and cost three hundred bucks to buy. Surely the publication process contains some kind of plausibility checks?
Whatever the story is, this article looks at key parameters in performance testing, the history, issues and suggests some practical solutions that could be considered in future amendments or editions. For the purpose of this article, the three standards will be called IEC1, IEC2 and AAMI to denote IEC 60601-2-24:1998, IEC 60601-2-24:2012 and ANSI/AAMI ID 26 (R)2013 respectively.
Minimum rates
In IEC1, the minimum rate is defined as being the lowest selectable rate but not less than 1mL/hr. There is a note that ambulatory use pumps should be the lowest selectable rate, essentially saying ignore the 1mL/hr for ambulatory pumps. OK, time to be pedantic - according to IEC rules, “notes” should be used for explanation only, so strictly speaking we should apply the minimum 1mL/hr rate. The requirement should have simply been “the lowest selectable rate, but not less than 1mL/hr for pumps not intended for ambulatory use”. It might sound nit-picking, but putting requirements in notes is confusing, and as we will see below, even the standards committee got themselves in a mess trying to deal with this point.
The reason 1mL/hr makes no sense for ambulatory pumps is that they usually have rates that are tiny compared to a typical bedside pump - insulin pumps for example have basal (background) rates as low as 0.00025mL/hr, and even the highest speed bolus rates are less than 1mL/hr. So a minimum rate of 1mL/hr makes absolutely no sense.
The AAMI standard tried to deal with this by deleting the note, creating a new defined term - “lowest selectable rate” and then adding a column to Table 102 to include this as one of the rates that can be tested. Slight problem, though, is that they forgot to select this item in Table 102 for ambulatory pumps, the very device that we need it for. Instead, ambulatory pumps must still use the “minimum rate”. But, having deleted the note, it meant that ambulatory pumps have to be tested 1mL/hr - a basal rate that for insulin would probably kill an elephant.
While making a mess of the ambulatory side, the tests for other pumps are made unnecessarily complex as they do require tests at the lowest selectable rate in addition to 1mL/hr. Many bedside pumps are adjustable down to 0.1mL which is the resolution of the display. However, no one really expects this rate to be accurate, it’s like expecting your car’s speedometer to be accurate at 1km/hr. It is technically difficult to test, and the graphs it produces are likely to be messy and meaningless to the user. Clearly not well thought out, and appears to be ignored by manufacturers in practice.
IEC2 also tried to tackle the problem. Obviously following AAMI’s lead, they got as far as deleting the note in the minimum rate definition, adding a new defined term (“minimum selectable rate”), … but that’s as far as it got. This new defined term never gets used in the normative part of the standard. The upshot is that ambulatory pumps are again required to kill elephants.
So what is the recommended solution? What probably makes the most sense is to avoid fixing it at any number, and leave it up to the manufacturer to decide a “minimum rate” at which the pump provides reliable repeatable performance. This rate should of course be declared in the operation manual, and the notes (this time used properly as hints rather than a requirement) can suggest a typical minimum rate of 1mL/hr for general purpose bedside pumps, and highlight the need to assess the risks associated with the user selecting a rate lower than the declared minimum rate.
That might sound a bit wishy washy, but the normal design approach is to have a reliable range of performance which is greater than what people need in clinical practice. In other words, the declared minimum rate should already be well below what is needed. As long as that principle is followed, the fact that the pump can still be set below the “minimum rate” is not a significant risk. Only if the manufacturer tries to cheat - in order to make nicer graphs - and select a “minimum rate” that is higher than the clinical needs that the risk starts to become significant. And competition should encourage manufacturers to use a minimum rate that is as low as possible.
Maximum rate
In IEC1, there are no performance tests required at the maximum selectable rate. Which on the face of it seems a little weird, since the rate of 25mL/hr is fairly low compared to maximum rates in the order of 1000mL/hr or more. Performance tests are normally done at minimum, intermediate and maximum settings.
AAMI must have thought the same, and introduced the term “maximum selectable rate”, and added another column to Table 102, requiring tests at the maximum rate for normal volumetric and other types of pumps with continuous flow (excluding ambulatory pumps).
Sounds good? Not in practice. The tests in the standard cover three key aspects - start up profile, short to medium term variability, and long term accuracy. Studying all three parameters at all four points (lowest selectable, minimum, intermediate and maximum selectable, i.e. 0.1, 1.0, 25, 999mL/hr) is something of a nightmare to test. Why? Because the range from represents 4 orders of magnitude. You can’t test that range with a single set up, you need at least two and possibly three precision balances with different ranges. A typical 220g balance that is used for IEC testing doesn’t have the precision for tests at 0.1mL/hr or the range for tests at 999mL/hr.
IEC2 again started to follow AAMI, adding the new defined term “maximum selectable rate”. But again, that’s pretty much as far as it got. It was never added to Table 201.102. Fortunately, unlike the other stuff ups, this one is benign in that it does not leave the test engineer confused or force any unrealistic test. It just defines a term that is never used.
The recommended solution? A rate of 25ml/hr seems a good rate to inspect the flow rate start up profile, trumpet curves and long term accuracy. As a pump gets faster, start up delays and transient effects will become less of an issue. This suggests that for higher speeds, the only concern is the long term flow rate accuracy. This can be assessed using fairly simple tests that use the same precision balance (typically 220g, 0.1mg resolution), over shorter periods. For example, an infusion pump with 1000mL/hr range could be tested at 100, 500 and 1000mL/hr by measuring the time taken to deliver 100mL, or any volume which is large enough that the measurement errors are negligible. Flow rate accuracy and pump errors can be easily be derived from this test, without needing multiple balances.
Sample intervals
IEC1 used a sample interval of 30s for general purpose pumps and 15min for ambulatory, both of which seem reasonable in practice.
AAMI changed this to 10s for general purpose pumps. The reason was probably to allow the true profile to be viewed at the maximum selectable rate, such as 999mL/hr. It also allows for a smoother looking trumpet curve (see below).
While that is reasonable, the drawback is that for low flow rates, the background “noise” such as resolution of the balance will appear three times larger, which is unrelated to the infusion pump’s performance. For example, a typical 220g balance with a 0.1mg resolution has a rounding error “noise” of 1.2% at rates of 1mL/hr sampled at 30s. If the sample interval is reduced to 10s, that noise becomes 3.6%, which is highly visible in a flow rate graph, is not caused by the pump and has no clinical relevance.
Needless to say, this is another AAMI deviation that seems to have been ignored in practice.
IEC2 again appears to have started to follow AAMI, and got as far as deleting the reference to a 30s interval (0.5 min). But, that’s as far as they got - the sentence now reads “Set the sample interval S”, which implies that S is somehow defined elsewhere in the standard. Newbie test engineers unaware of the history could spend hours searching the standard to try and find where or how S is determined. A clue that it is still 0.5 min can be found in Equation (6) which defines j and k as 240 and 120, which only makes sense if S = 0.5 min. With the history it is clear that there was an intention to follow AAMI, but cooler heads may have prevailed and they just forgot to re-insert the original 0.5min.
Recommended solution? The 30s interval actually makes a bumpy looking flow graph, so it would be nice to shift to 10s. The “noise” issue can be addressed by using a running average over 30s, calculated for each 10s interval. For example, if we have weight samples W0, W1, W2, W3, W4, W5, W6 covering the first minute at 10s intervals, the flow rate data can be plotted every 10s using data derived from (W3-W0), (W4-W1), (W5-W2), (W6-W3). That produces a smoother looking graph while still averaging over 30s. It can also produce smoother looking trumpet curves as discussed below.
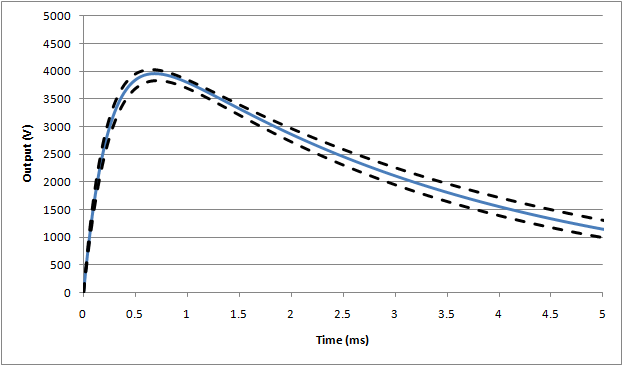
Trumpet curves
For general pumps, IEC1 requires the analysis to be performed at 2, 5, 11, 19 and 31 min (with similar points for ambulatory adjusted for the slower sample rate). That requires some 469 observation windows to be calculated, with 10 graph points.
The AAMI puts this on steroids: first it introduces a 10s sample interval, next it expands the range down to 1min, and finally requires the calculation to be made for all available observation windows. That results in some 47965 window calculations with 180 graph points
That said, the AAMI approach is not unreasonable: with software it is easy to do. It is possible to adopt a 10s sample interval from which a smooth trumpet curve is created (from 1 to 31 min as suggested). And, it is unclear why the IEC version uses only 2, 5, 11, 19 and 31min. It is possible that some pumps may have problem intervals hidden by the IEC approach.
So, somewhat unusually in this case, the recommended solution could be to adopt the AAMI version.
The balance (update 2019/09/20)
In figure 201.104b of IEC2, the balance in the diagram is shown as having four decimal places, while the note below says that a balance “accurate to” 5 decimal places “is required” for pumps with low minimum rates. This has a bunch of issues detailed below.
First, IEC/ISO Directive 2, Clause 28.5.4 “Notes to figures shall not contain requirements or any information considered indispensable for the use of the document”. So, again the writers have failed to follow the rules of writing standards.
Next, the note states that the balance should be “accurate to 5 decimal places”. It is most likely that the writers meant a resolution of 5 decimal places. To be accurate to 5 decimal places normally implies that the actual device displays an extra digit beyond, in other words it would require a balance that displays a resolution down to 0.000000g. Good luck finding that. Just to be pedantic (again) the note is missing any units. We assume it is grams based on the diagram, but …
Finally, ignoring the above issues and assuming the writers meant a resolution of 5 decimal places - research indicates that scales with a range of 220g and a resolution of 5 digits do exist, but they are extremely expensive and the specifications are not great. Noting that 0.00001g is effectively a resolution of 45 ppb (parts per billion) it looks like the balance manufacturers can achieve that resolution only with averaging, which could potentially affect the results. The scales would also be highly susceptible to vibration, and finally finding a calibration laboratory that is willing to check the device at that level of precision is likely to be very expensive if not impossible.
AAMI deleted this note. That says it all.
Back pressure
IEC1 and IEC2 require tests at the intermediate rate to be performed using a back pressure of ±100mmHg. This is easily done by shifting the height of the pump above or below the balance (approximately 1.5m = 100mmHg).
AAMI decided to change this to +300mmHg / -100mHg. The +300mmHg makes some sense in that real world back pressures can exceed 100mmHg. However, it makes the test overly complicated: it is likely to involve mechanical restriction which will vary with time and temperature and hence needs to be monitored and adjusted during tests up to 2hrs.
Recommended solution: The impact on accuracy is likely to be linearly related to pressure, so for example if there is a -0.5% change at +100mmHg, we can expect a -1.5% change at +300mmHg. The IEC test (±100mmHg) is a simple method and gives a good indication if the pump is influenced by back pressure, and allows direct comparison between different pumps. Users can estimate the error at different pressures using this value. Hence, the test at ±100mmHg as used in the IEC standards makes sense.
Update 2019/09/20: it has been pointed out that the above assumption of linearity between pressure and flow may not be valid. This suggests more work is required to investigate the effects and to develop a repeatable set up if higher pressures are required.
Pump types
In IEC1 and AAMI, there are 5 pump types in the definition of an infusion pump. The types (Type 1, Type 2 etc) are then used to determine the required tests.
In IEC2, the pump types have been relegated to a note, and only 4 types are shown. The original Type 4 (combination pumps) were deleted, and profile pumps, formally Type 5 are now referred to as Type 4. But in any case … it’s a note, and notes are not part of the normative standard.
Unfortunately this again seems to be a case of something half implemented. While the definition was updated the remainder of the standard continues to refer to these types as if they were defined terms, and even more confusingly, referring to Types 1 to Type 5 according to the definition in the previous standard. A newbie trying to read the standard would be confused by the use of Type 5 which is completely undefined, and by the fact that references to Type 4 pumps may not fit with the informal definition. A monumental stuff up.
In practice though the terms Type 1 to Type 5 were confusing and made the standard hard to read. It seems likely that the committee may have relegated it to a note with the intention of replacing the normative references with direct terms such as “continuous use”, “bolus”, “profile” and “combination” and so on. So for practical terms, users might just ignore the references to “Type” and hand write in the appropriate terms.
Drip rate controlled pumps
Drip rate controlled pumps were covered in IEC1, but appear to have been removed in IEC2. But … not completely - there are lingering references which are, well, confusing.
A possible scenario is that the authors were planning to force drip rate controlled pumps to display the rate in mL/hr by using a conversion factor (mL/drip), effectively making them volumetric pumps, and allowing the drip rate test to be deleted. The authors then forgot to delete a couple of references to “drop" (an alternative word to “drip”), but retained the test for tilting drip chambers.
Another possible explanation is that the authors were planning to outlaw drip rate controlled pumps altogether, and just forgot to delete all references. That seems unlikely as parts of the world still seem to use these pumps (Japan for example).
For this one there is no recommended solution - we really need to find out what the committee was thinking.
Statistical variability
Experience from testing syringe pumps indicates there is a huge amount of variability to the test results from one syringe to another, and from one brand to another, due to variations in “stiction”: the stop start motion of the plunger. And even for peristaltic type pumps the accuracy is likely to be influenced by the tube’s inner diameter, which is also expected to vary considerably in regular production. While this has little influence on the start up profile and trumpet curve shape, it will affect the overall long term accuracy.
Nevertheless, the standard only requires the results from a single “type test” to be put in the operation manual, leading to the question - what data should I put in? As it turns out, competitive pressures are such that manufacturers often use the “best of the best” data in the operation manuals, which is totally unrepresentative of the real world. Their argument is that - since everybody does it, … well … we have to do it. And, usually the variability is coming from the administration set or syringe, which the pump designers feel they are not responsible for.
In all three standards this issue is discussed (Appendix AA.4), but it is an odd discussion since it is disconnected with the normative requirements in the standard: it concludes that for example, a test on one sample is not enough, yet the normative parts only requires one sample.
Recommended solution (updated 2019/09/20)? Given that pumps, syringes and administration sets are placed on the market separately, it makes sense that separate standards (or separate sections in the standard) are developed for each part of the system. For example, tests could be developed to assess the variability of stiction in syringes with minimum requirements and declarations of performance if they are to be used with syringe pumps. Competitive pressure would then encourage manufacturers to work on making smoother action at the plunger.
Regulatory status
One might wonder why IEC 60601-2-24:2012 (EN 60601-2-24:2015) is yet to be adopted in Europe. In fact there are quite a few particular standards in the IEC 60601 series which have yet to be adopted in Europe. One possible reason is that the EU regulators are starting to having a close look at these standards and questioning whether they do, indeed, address the essential requirements. Arguably EN 60601-2-24:2015 does not, so perhaps there is a deliberate decision not to harmonise this and other particular standards that have similar problems.
The FDA , mysteriously, has no reference to either IEC and AAMI versions - mysteriously because it is a huge subject with lots of effort by the FDA to reduce infusion pump incidents, yet references to standards either formally or in passing seem to be non-existent.
Health Canada does list IEC 60601-2-24:2012, but interestingly there is a comment below the listing that “Additional accuracy testing results for flow rates below 1 ml/h may be required depending on the pump's intended use”, suggesting that they are aware of short comings with the standard.
Overall conclusion
Ultimately, it may be that the IEC committees are simply ill-equipped or structurally unsound to provide particular standards for medical use. The IEC is arguably set up well for electrical safety, with heavy representation by test laboratories and many, many users of the standards - meaning that gross errors are quickly weeded out. But for particular standards in the medical field, reliance seems to be largely placed on relatively few manufacturers, with national committees unable to provide plausible feedback and error checking. The result is standards like IEC 60602-2-24:2012 - full of errors and questionable decisions with respect to genuine public concerns. It seems we need a different structure - for example designated test agencies with good experience in the field, that are charged with reviewing and “pre-market” testing of the standard at the DIS and FDIS stages, with the objective of improving the quality of particular standards for medical electrical equipment.
Something needs to change!

 Introduction
Introduction
.png)